베르시포로신, 폐섬유화 유발하는 콜라겐 합성을 직접 억제하는 차별화된 효능
목표 인원 약 60% 달성으로 임상 순항…2025년까지 2상 완료 계획
[서울=뉴스핌] 이나영 기자= 대웅제약은 세계 최초 신약(First-in-class)으로 개발 중인 특발성 폐섬유증 치료제 '베르시포로신(DWN12088)'이 임상 2상에서 안전성을 검증 받아 상용화에 한 걸음 더 다가섰다고 29일 밝혔다.
베르시포로신은 지난 3월 개최한 1차 독립적 데이터 모니터링 위원회(Independent Data Monitoring Committee, 이하 IDMC) 회의에 이어, 7월 26일 개최한 2차 회의에서도 임상 지속을 권고 받았다. 이번 2차 IDMC 회의에서는 임상시험을 완료한 특발성 폐섬유증 환자 51명을 포함한 총 59명의 등록 환자를 대상으로 베르시포로신의 안전성 데이터를 심층 검토한 결과 큰 문제점이 발견되지 않았다.
IDMC는 내년 초 예정된 3차 회의에서 베르시포로신 임상 2상의 안전성을 최종 점검할 예정이다. 임상 2상은 2025년 내로 완료할 계획이다.
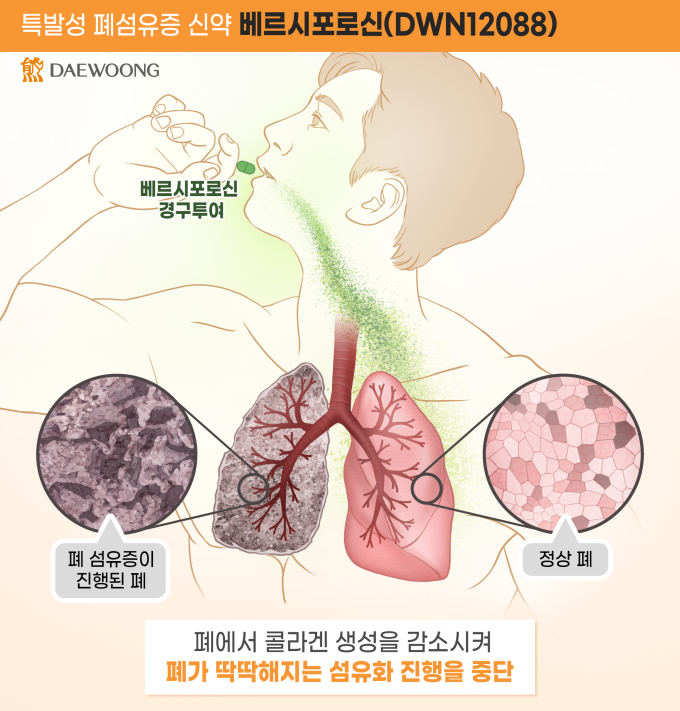
특발성 폐섬유증(Idiopathic Pulmonary Fibrosis, IPF)은 폐에 콜라겐이 비정상적으로 축적되어 폐 기능이 상실되는 난치병으로, 진단 후 5년 생존율이 40%에 불과할 정도로 예후가 좋지 않은 치명적 질환이다. 기존 치료제는 섬유화 진행의 속도를 늦추는 수준으로 효능이 매우 제한적이고, 이상 반응의 발생률도 높다.
베르시포로신은 콜라겐 합성을 직접적으로 억제하는 새로운 작용 메커니즘을 통해 기존 치료제와 차별화된 안전성과 효능을 보여줄 것으로 기대된다. 임상 1상에서 건강한 사람을 대상으로 안전성과 약동학적 특성을 확인했다. 임상 2상은 40세 이상의 특발성 폐섬유증 환자를 대상으로 진행되며, 현재 허가된 치료제를 복용 중이거나 중단한 환자들이 참여하고 있다.
임상시험은 24주 동안 진행되며, 베르시포로신 단독 및 기존 치료제와 병용 요법의 안전성, 내약성, 유효성을 평가한다. 2023년 1월 미국과 한국에서 시작된 임상 2상은 현재까지 61명의 환자를 모집하여 목표 인원 102명의 약 60%를 달성하는 등 순항 중이다. 국내 임상시험은 서울아산병원, 신촌세브란스병원, 서울삼성병원, 순천향대 부천병원, 부천성모병원, 아주대병원, 명지병원, 울산대병원, 인제대 부산백병원 등 10곳에서 수행되고 있다.
이창재 대웅제약 대표는 "이번 IDMC의 권고는 베르시포로신의 원활한 개발에 있어 안전성을 입증한 중요한 이정표"라며, "혁신 신약 후보 물질인 베르시포로신의 개발을 통해 특발성 폐섬유증 환자들에게 새로운 치료 옵션을 제공할 수 있도록 최선을 다하겠다"고 밝혔다.
한편, 베르시포로신은 미국 FDA로부터 2019년 희귀의약품 및 2022년 신속심사제도(패스트 트랙) 개발 품목으로 지정 받았으며, 올해 1월에는 유럽의약품청(European Medicines Agency, EMA)으로부터 희귀의약품으로 지정된 바 있다. 지난 2023년에는 영국의 씨에스파마슈티컬스(CS Pharmaceuticals)와 중화권 기술 수출 계약을 체결하는 등 글로벌 시장 진출에 박차를 가하고 있다.
nylee54@newspim.com